Intro 2: Explanation of observational and antibody kinetics models
David Hodgson
2026-03-03
model_define.RmdIntroduction
This document explains two critical parts of the model defined in the provided R code:
- The Observational Model: This component models the observed data, specifying how the measured values relate to the latent (unobserved) processes.
- The Antibody Kinetics Model: This component models the changes in antibody levels over time, accounting for both natural decay (waning) and infection-induced changes.
1. The Observational Model
Definition
The observational model captures the process by which observed data
(IgG in this case) is generated from the underlying system.
It defines the relationship between the true (latent) variables and the
observed data.
Code Breakdown
obsLogLikelihood <- function(titre_val, titre_est, pars) {
ll <- dnorm(titre_val, titre_est, pars[1], log = TRUE)
ll
}
observationalModel <- list(
names = c("IgG"),
model = makeModel(
addObservationalModel("IgG", c("sigma"), obsLogLikelihood)
),
prior = bind_rows(
addPrior("sigma", 0.0001, 4, "unif", 0.0001, 4)
)
)names = c("IgG"): The observed biomarker here isIgG, representing some measurement, such as a biomarker (e.g., antibody concentration).model = makeModel(...): The model is created using themakeModelfunction, which defines how the observation model is constructed.addObservationalModel("IgG", c("sigma"), obsLogLikelihood): This line adds an observational model forIgG, where the parametersigmais used to model the noise or uncertainty in the observations. The log-likelihood function (obsLogLikelihood) is used to define how the observations relate to the latent variable.prior = bind_rows(...): This block defines the prior distribution for the parametersigma, which controls the observation noise.addPrior("sigma", 0.0001, 4, "unif", 0.0001, 4): The parametersigmais given a uniform prior distribution between0.0001and4, indicating that the model expects a certain range of variability in the observed data.
2. The Antibody Kinetics Model
Definition
The antibody kinetics model describes how antibody levels evolve over time in response to different factors, such as infection or natural waning. This model accounts for both natural decay in antibody levels and changes due to infection.
Code Breakdown
Define the antibody kinetic functions
noInfSerumKinetics <- function(titre_est, timeSince, pars) {
titre_est_new <- titre_est - (pars[1] * titre_est) * (timeSince)
titre_est_new <- max(titre_est_new, 0)
titre_est_new
}
infTuenisPower2016 <- function(titre_est, timeSince, pars) {
y1 <- pars[1]
t1 <- pars[2]
r <- pars[3]
alpha <- pars[4]
v <- 0.001
mu <- 1 / t1 * y1
if (timeSince < t1) {
titre_est_boost <- exp(mu * timeSince)
} else {
titre_est_boost <- exp(y1) * (1 + (r - 1) * exp(y1)^{r - 1} * v * (timeSince - t1)) ^ {-1 / (r - 1)}
}
titre_est_log <- titre_est + log(titre_est_boost) * max(0, 1 - titre_est * alpha)
titre_est_log
}Create the antibody kinetics model
abkineticsModel <- list(
model = makeModel(
addAbkineticsModel("none", "IgG", "none", c("wane"), noInfSerumKinetics),
addAbkineticsModel("inf", "IgG", "inf", c("y1_h1", "t1_h1", "r_h1", "s"), infTuenisPower2016)
),
prior = bind_rows(
addPrior("y1_h1", 1, 6, "unif", 1, 6),
addPrior("t1_h1", 3, 14, "unif", 3, 14),
addPrior("r_h1", 1, 5, "unif", 1, 5),
addPrior("s", 0, 1, "unif", 0, 1),
addPrior("wane", 0, 1, "unif", 0, 1)
)
)model = makeModel(...): The antibody kinetics model is built usingmakeModel, which defines two different antibody response models.addAbkineticsModel("none", "PreF", "none", c("wane"), noInfSerumKinetics): This line models the antibody kinetics when there is no infection. The “PreF” biomarker is modeled with the parameterwane, which represents the natural decay or waning of antibody levels over time. ThenoInfSerumKineticsfunction describes this process.-
addAbkineticsModel("inf", "PreF", "inf", c("y1_h1", "t1_h1", "r_h1", "s"), infTuenisPower2016): This line models the antibody kinetics after infection. Several parameters describe the response to infection:-
y1_h1: Initial antibody level post-infection. -
t1_h1: Time of peak response. -
r_h1: Rate of antibody decay post-infection. -
s: Slope of decay after peak response. -
infTuenisPower2016: The function governing the infection-induced antibody response.
-
-
prior = bind_rows(...): This block defines prior distributions for the model parameters:-
y1_h1: Uniform prior between1and6, representing the initial antibody level after infection. -
t1_h1: Normal prior with mean14and standard deviation3, representing the time to peak response. -
r_h1: Uniform prior between1and5, representing the rate of antibody decay. -
s: Uniform prior between0and1, representing the slope of the decay post-infection. -
wane: Uniform prior between0and1, representing the natural waning of antibody levels over time.
-
Summary
The antibody kinetics model accounts for the natural waning of
antibodies in the absence of infection, as well as the antibody response
after infection. Parameters such as y1_h1,
t1_h1, and r_h1 govern the kinetics of
antibody production and decay. The model uses both uniform and normal
priors to specify reasonable ranges for these parameters.
3. Full model definition
This R code defines a serological model using different components
such as biomarkers, exposure types, observational and antibody kinetics
models, and priors. The code creates a model definitions
(modeldefinition) and uses these to create a serological
model for data analysis (seroModel).
Step-by-Step Breakdown
Step 1: Define the First Model modeldefinition
Once all the data is loaded in, the first step is to define the
model. This includes the biomarkers, exposure types, exposure fitted,
observational model, and antibody kinetics model. Then, once the list is
created, the createSeroJumpModel function is used to create
the serojump model.
# See data_format.Rmd vignette about data input types
biomarkers <- "IgG"
# Create the data_sero dataframe
data_sero <- data.frame(
id = c(1, 1, 2, 2, 3, 3),
time = c(1, 50, 1, 50, 1, 50),
IgG = c(1.2, 4.4, 1.2, 4.4, 3.0, 3.0)
)
# Define possible exposure types
exposureTypes <- c("none", "inf")
# Create the exposure_data dataframe
exposure_data <- data.frame(
id = c(1),
time = c(14),
exposure_type = c("inf")
)
exposureFitted <- "inf"
# Create the attack_rate_data dataframe
attack_rate_data <- data.frame(
time = rep(1:25),
prob = rep(1/25, 25)
)
seroModel <- createSeroJumpModel(
data_sero = data_sero,
data_known = NULL,
biomarkers = biomarkers,
exposureTypes = exposureTypes,
exposureFitted = exposureFitted,
observationalModel = observationalModel,
abkineticsModel = abkineticsModel)## OUTLINE OF INPUTTED MODEL
## There are 1 measured biomarkers: IgG
## There are 2 exposure types in the study period: none, inf
## The fitted exposure type is inf
## PRIOR DISTRIBUTIONS
## Prior parameters of observationalModel are: sigma
## Prior parameters of abkineticsModel are: y1_h1, t1_h1, r_h1, s, wane
## No single entries in data_sero!,
## No individuals with less than 15 days in the study!
## Exposure rate is not defined over the time period. Defaulting to uniform distribution between 1 and 50 .4.Pre run sanity plots
Before running the whoel model it is good to check the data and the
priors. This can be done using a suit of functions
plotPriors function.
p1 <- plotSero(seroModel)
p2 <- plotPriorPredictive(seroModel)## Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
## ℹ Please use `linewidth` instead.
## ℹ The deprecated feature was likely used in the serojump package.
## Please report the issue to the authors.
## This warning is displayed once per session.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.
p3 <- plotPriorInfection(seroModel)
p1 / p2## Ignoring unknown labels:
## • colour : "Exposure type"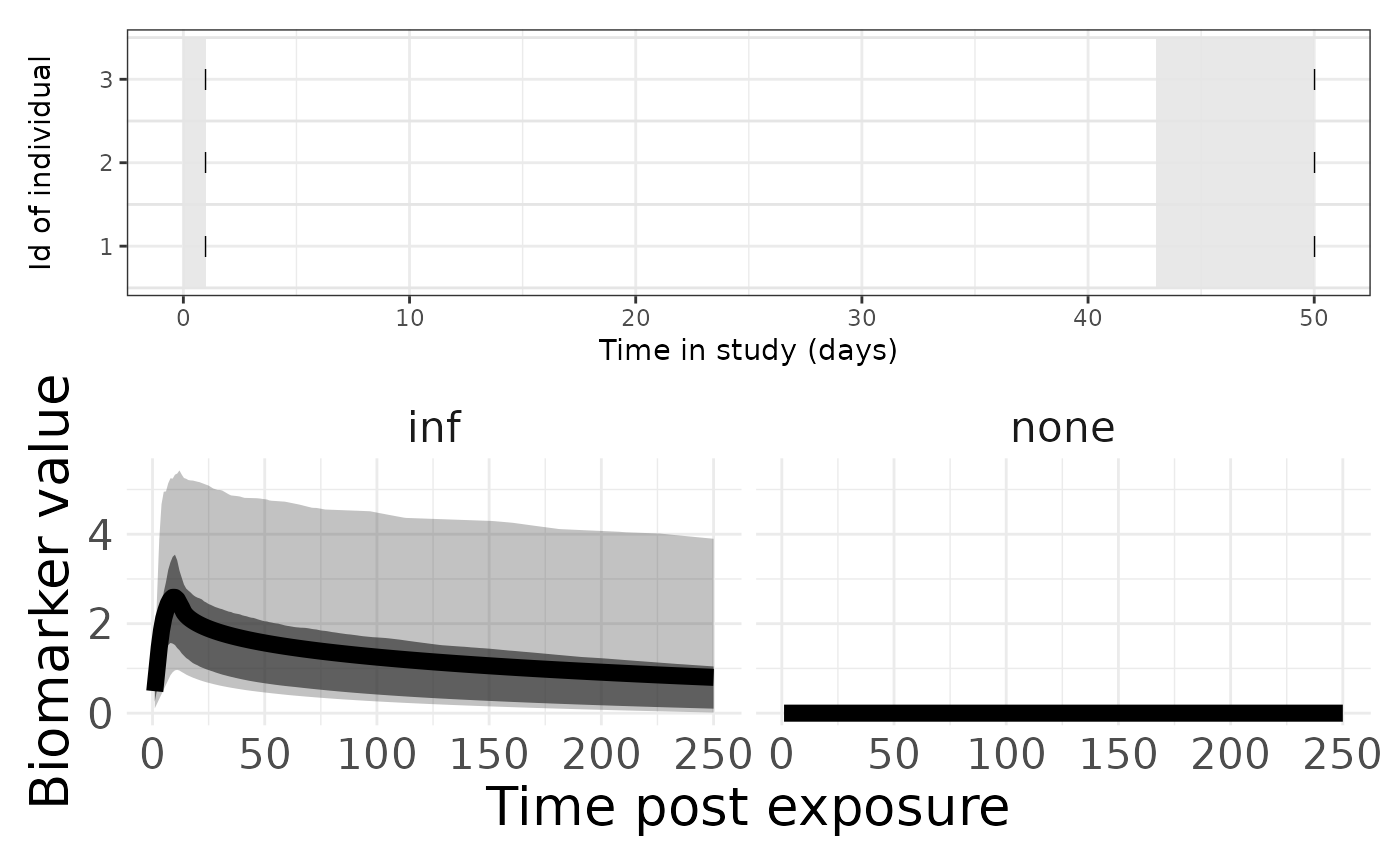
p3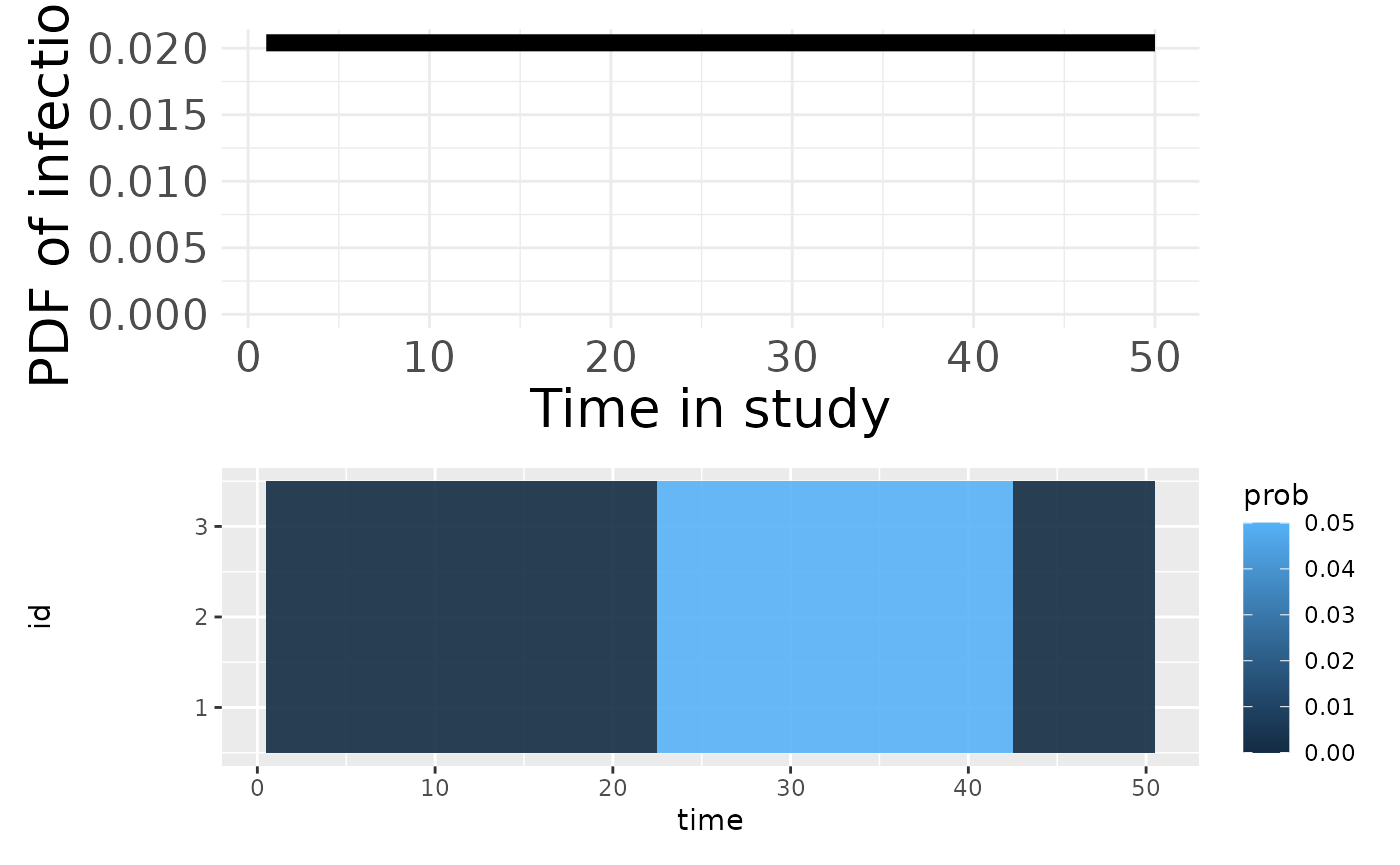
4. Run the model!
Once seroModel is defined, the user can run the model using the
runSeroJump function. This function requires the seroModel,
settings for the RJ-MCMC algorithm, and information about saving the
model outputs.
save_info <- list(
file_name = "simple",
model_name = "ex1"
)
rj_settings <- list(
numberChainRuns = 4,
numberCores = mc.cores,
iterations = 2000,
burninPosterior = 1000,
thin = 1
)
set.seed(100)
model_summary <- runSeroJump(seroModel, rj_settings, save_info = save_info)5. Plot the outputs
# Need to have save_info model_summary to run these
plotMCMCDiagnosis(model_summary, save_info = save_info) # plots the convergence diagnosis
plotPostFigs(model_summary, save_info = save_info) #plots some plots of the posteriorsmodel_summary$post has all the posterior distributions
which the user can extract and plot themselves!